筆者:Vishal Chakravarty
ブレグジットは医薬品規制を「なくした」のではありません。規制を「二重化」しました。
英国の製薬業界は2021年1月1日、EUで統一されていた規制枠組みから、英国(UK)とEUの双方で並行して対応が必要な「二分化したコンプライアンス環境」へ移行しました。2025年1月1日に実施されたウィンザー・フレームワークは、北アイルランド(Northern Ireland)固有の一部の複雑さを解消した一方で、「UKとEUの二重インフラを維持する必要がある」という根本的な現実を変えたわけではありません。
これは単なる事務負担ではありません。欧州の医薬品流通における競争力を変える「構造コスト」です。
共有から分離へ:コンプライアンスの転換
ブレグジット以前、英国の製薬企業はEUの規制調和の中で事業を行っていました。GMP(Good Manufacturing Practice:適正製造基準)査察、バッチリリース、ファーマコビジランス(PV)報告、サプライチェーン文書などは、加盟国全体で運用される共通基準に沿っていました。英国に拠点を置くQualified Person(QP:製造販売承認に基づく出荷判定の責任者)がEU向けバッチの認証を行うことも可能で、Pharmacovigilance System Master File(PSMF:PVシステム・マスターファイル)も単一で両法域をカバーし得ました。トレーサビリティも欧州全体のリポジトリと連携していました。
ブレグジット後、この統合は崩れました。EUの規制上、英国は「第三国」となり、UKとEUで別々に走るコンプライアンス義務が並行稼働することになりました。
QP要件:地理的分離がもたらす影響
運用面で最も影響が大きい変更の一つが、バッチリリリースにおけるQP認証です。
EU向け製品のバッチは、EEA(European Economic Area:欧州経済領域)加盟国に物理的に所在するQPによって認証される必要があります。英国に拠点を置くQPは、EU流通向けの認証を担えません。一方、UK市場向けのバッチリリースには、UK側の要件に沿ったUK拠点でのQP体制が必要になります。
この地理的分離は、運用上、次のような含意をもたらします:
・英国企業がEUへ供給する場合:EEA内でQP体制を確保する必要があります。QP機能の移転、EU側のCMO(受託製造機関)活用、あるいは適切なGMP設備・人材を備えた子会社体制の構築などが必要になります。
・EU企業が英国へ供給する場合:逆方向でも同様です。EUのQPだけではUK向けリリースを担えず、UK側のQP体制を整えるか、UK拠点のパートナーを活用する必要があります。
・臨床試験(治験薬):2022年1月以降、グレートブリテン(GB)内の治験施設に入る治験薬について、UK側のQP監督が求められるなど、UK/EUに跨る試験ではプロセス変更が必要になっています。
また、二重QPインフラを整備するための猶予は大きくありませんでした。移行期間(2020年12月31日)までにQP認証体制が更新されていない場合、製品や手続によっては、当該市場におけるコンプライアンス制約や、適法なバッチリリリースへの影響が生じ得ます。
バッチ試験・出荷判定プロトコル
QP要件と密接に関係するのが、バッチ試験の場所と出荷判定(release)手続です。
従来、英国の試験施設がMarketing Authorizations(販売承認)の記載サイトとして認められている場合、UK側で試験を実施し、UKのQP確認でEUリリースに繋げる運用もあり得ました。しかしブレグジットにより、この相互的な認定は終了しました。
現在の枠組みでは、概ね次のような対応が必要になります:
・EU市場向け:EEA域内の試験サイト、または英国がもはや保持しない相互承認協定(MRA)対象施設等での試験が必要になります。移行措置の名残でUKサイトがEUの承認書に残っている場合でも、EUへの輸入試験が求められる可能性があります。EUのQPは、特定の貿易協定上の根拠がない限り、UKのQP確認のみを根拠にできません(そのような取り決めは限定的です)。
・UK市場向け:UK側の基準(MHRA)に沿った試験・QP認証が必要になります。
結果として、同一製品でもUK向け/EU向けで、別々の地域・別々のQPプロセスが必要となり、運用が重複します(Figure 1参照)。
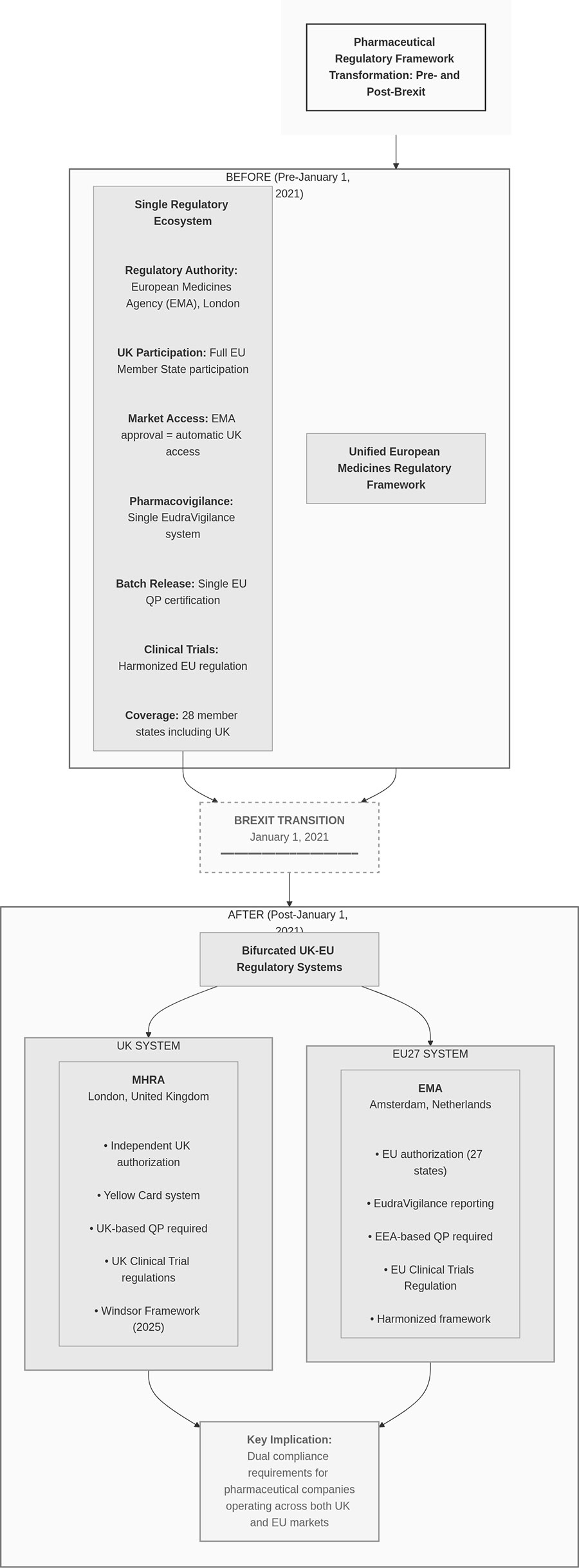
Figure 1. 制度のBefore/After比較(コンプライアンス運用)
※画像クリックで拡大表示
ファーマコビジランス(PV)体制の分離
PVインフラも同様に二分化しました。
▽PSMF(Pharmacovigilance System Master File)
UKの販売承認(GBおよび北アイルランドを含む)では、MHRAから求められた際に提示できるPSMFを維持する必要があります。単一のPSMFでUK承認製品全体をカバーできる場合でも、疑われる副作用情報にアクセスできるUK拠点と同一地点から電子的にアクセス可能であることが求められます。
また、UKのPVシステムごとにUK PSMF番号がMHRAにより付与されます(付与の処理期間は運用により変動し得ます)。
北アイルランドに関わる製品では、ウィンザー・フレームワークに伴う特殊性から、PV活動の主要拠点やQPPV(Qualified Person for Pharmacovigilance:PV責任者)の所在に応じて、PSMFの位置づけ・配置の考慮が必要になります。
▽UK QPPV(UK Qualified Person for Pharmacovigilance)
UK QPPVはEU GVP(Good Pharmacovigilance Practices)と類似した要件で運用され、医学的専門性(または医学的知見へのアクセス)が求められます。UK QPPVはUKまたはEEAに居住し得ますが、UKで承認された製品のPV体制に対して正式責任を負います(法的責任はUKのMAH:Marketing Authorization Holderが負います)。
▽有害事象報告(副作用報告)
UKの報告はEudraVigilanceではなく、MHRAのYellow Cardシステムに流れます。両法域で事業を行う企業は、データフローや期限、シグナル検出を含む二重の報告体制を維持する必要があります。
この分離は、同一製品の安全性データについて、UK当局向けとEU当局向けの報告の切り分け(segregation)と、グローバル安全性監視としての統合(integration)の両立を求めます。
偽造医薬品指令(FMD):UKでの適用除外
2025年1月1日以降、EUの偽造医薬品指令(FMD:Falsified Medicines Directive)の安全機能に関する扱いは、UKで大きく分岐しました。
▽ウィンザー・フレームワーク以前:FMDはEUおよび北アイルランドに適用され、ユニークIDを含む2Dバーコードや改ざん防止包装が求められ、供給網の中でスキャン・デコミッショニングされていました。
▽ウィンザー・フレームワーク以後:2025年1月1日以降、FMDは北アイルランドを含むUK全域で適用されません。UK向け医薬品の包装にFMD準拠の安全機能を表示してはならず、2Dバーコードやシリアルが存在する場合も、欧州のリポジトリシステムで認識されないことが必要です。FMD準拠コードが存在する場合は除去または被覆が必要です。
補足として重要なのは、要点が「EUのリポジトリにアップロードされたFMDのユニークIDが除去/被覆され、EU側で認識されない状態にする」ことにある、という点です。
この結果、EU向けにはFMD要件が必要である一方、UK向けには「機能するFMD準拠」が成立しないようにしなければならない、というラベリング分岐が生じます(Figure 2参照)。
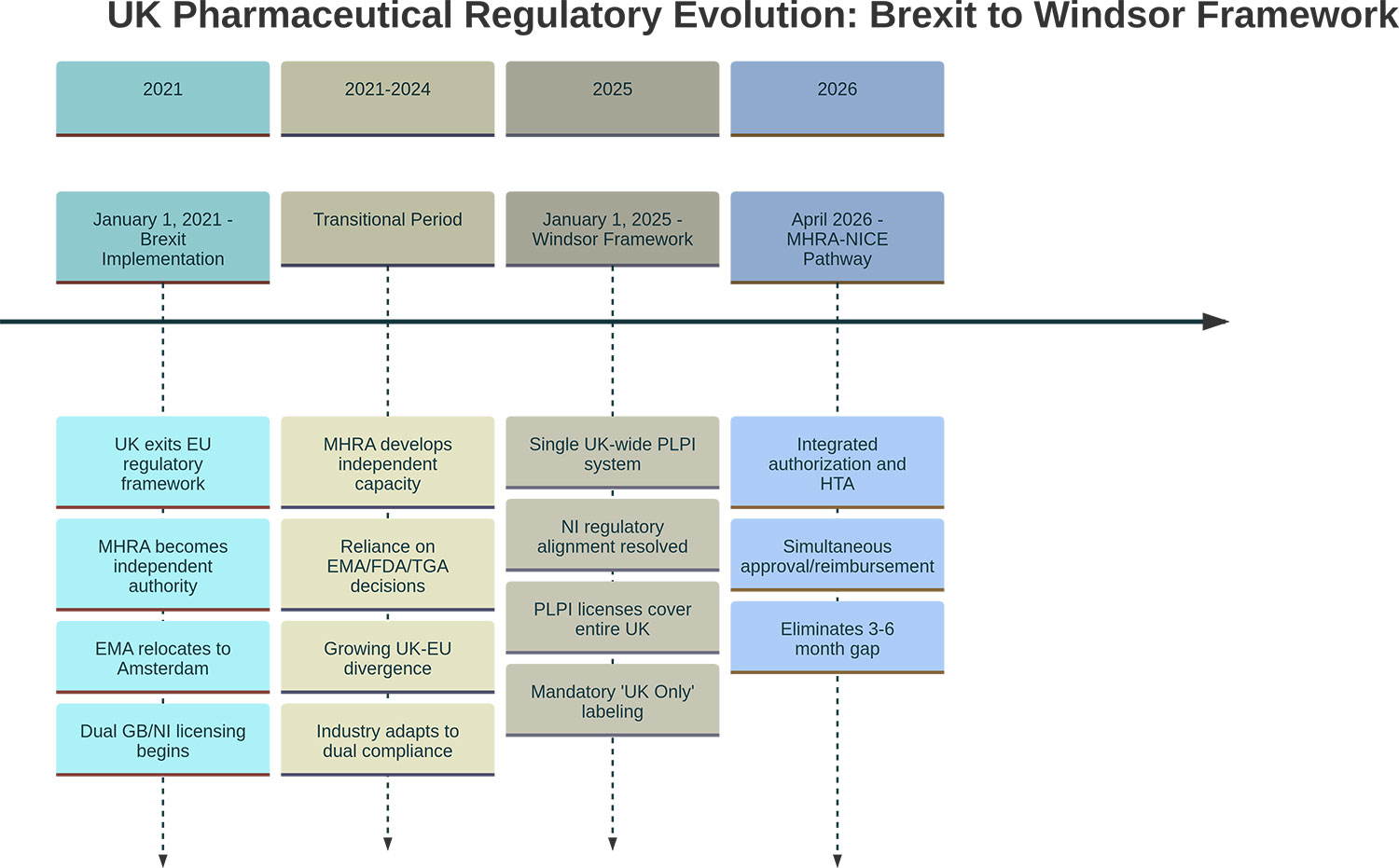
Figure 2.
※画像クリックで拡大表示
(図注)
・EMAのアムステルダム移転は2019年3月に実施。
・MHRA×NICEのAligned Pathwayは2025年10月に早期導入が開始。統合的なJoint Scientific Adviceは2026年4月までの開始が示されています。
表示要件:「UK Only」マーキング
2025年1月1日以降、UKで流通する医薬品は「UK Only」表示が求められます。この要件は並行輸入、卸流通、国内製造を含むUK市場に入る医薬品全般に及びます。
この表示は、UKとEUの流通経路の明確な切り分け、規制上の地位が異なる製品の越境移動の抑止、ウィンザー・フレームワークの遵守を可視化する目的を持ちます。
並行輸入の改定プロセスで輸入する卸業者は、2024年12月31日以降にQP認証された製品がMHRAのガイダンスに適合し、「UK Only」表示を備えることを確実にする必要があります。包装印字(printer)の適合性確認も求められます。
GDP:文書管理とトレーサビリティ
UKのGDP(Good Distribution Practice:適正流通基準)はEU GDPと原則として整合していますが、監督主体が分かれたことで、運用上は別々の監督下でコンプライアンスを実装する必要があります。
▽UK GDPの監督:MHRAが独立してGDPを運用します。EUの運用と異なり、MHRAは査察後の指摘(post-inspection letter)に対する回答をレビューし受理した後にGDP証明書を発行するため、証明書の発行プロセスやタイムラインが若干異なります。
▽文書要件:両制度で共通して、温度モニタリング(連続記録)、輸送手段の妥当性確認と輸送バリデーション、供給者の適格性確認と監査記録、製品の追跡可能性(メーカーから患者まで)、品質欠陥対応(原因究明を含む)など、包括的な文書管理が求められます。
両法域に供給する企業は、原則として同じ方向性の要件であっても、様式や保存要件などで法域固有の差分が出る可能性を織り込んだ文書体系が必要になります。
▽監査対応:UK側のQPサインオフ、文書のアクセス性、トレーサビリティ体制は、EUとは別のUK固有のタイムラインと手続に沿うMHRA査察に耐える状態(audit-ready)である必要があります。
サプライチェーン運用の調整
コンプライアンスの分離は、サプライチェーン上の実務調整を必要にしました:
・在庫管理:UK向け/EU向けで、表示要件(UK Only、FMD安全機能の有無)が異なるため、在庫の一体運用が難しくなり、柔軟性低下と運転資本の増加に繋がります。
・品質システム:UK/EUで別々のQP認証プロセスに対応するため、QMSを二重化するか、統合システム内で厳密に分離された運用が必要になります。
・規制インテリジェンス:MHRAとEMAの更新を別々に継続監視する必要があります。規制は必ずしも同時に更新されず、UK固有の事情でMHRAが独自に動く可能性があります(例:2025年7月の分散型製造関連の動き)。
・供給者管理:第三国(UK/EU以外)サプライヤーについて、UKとEUで別々の適格性評価が必要になる場合があります。
コスト構造への影響
統一から二分化への転換は、複数の次元で構造コストを押し上げます:
・人員:二重のQP体制、QPPV体制、MHRAとEMAに対応する独立した薬事体制。
・拠点:QP認証は管轄内での体制確保が前提となるため、UKとEEA双方での拠点整備または外部パートナー活用が必要になります。
・システム:PVデータベース、GDP文書管理、品質ワークフローなどの並行運用。
・表示・包装:UK Only表示とFMD要件差分に対応するための別ロット生産、あるいは製造後の変更工程。
これらのコストは企業規模によって吸収力が異なります。大手は高ボリュームの中で吸収しやすい一方、中小企業、とくにニッチ/スペシャリティ領域では相対的に負担が重く、市場参入判断に影響し得ます。
結果として、二重インフラを維持できる大規模組織が構造的に有利となり、小規模事業者が両市場同時展開を見送る要因になり得ます。
長期的コンプライアンスへの備え
UKの規制環境は、EUの合意を待たずに独自に進化し続けています。MHRAはEUのコンセンサスなしに規制上の革新を実装でき、MHRA–NICEのAligned Pathwayや、2025年7月に導入された分散型製造関連の動きはその例です。
こうした独自進化は、再統合ではなく、分岐の継続を意味します。両市場で事業を行う企業は、規制インテリジェンスの継続、法域差分に適応できる柔軟なコンプライアンス基盤、そしてUKとEUの要件が異なる場合にどの革新を採るかという戦略判断が求められます。
内部の品質システムを強化し、両法域の専門性へ投資し、独立進化に追随できる柔軟なコンプライアンス基盤を構築できる組織ほど、この二分化環境で持続的な事業運営を行いやすくなります。
ブレグジット後のコンプライアンス課題は一時的な移行対応ではありません。欧州の医薬品規制環境における恒久的な構造変化です。
Author Profile(著者プロフィール)

氏名(Name):Vishal Chakravarty
役職(Title):Founder & CEO, NovaPharm Healthcare Ltd
連絡先(E-mail):vishal@novapharmhealthcare.com
企業ウェブサイト:https://novapharmhealthcare.com/
執筆ポートフォリオ:https://vishal.novapharmhealthcare.com/
Bio(略歴)
Vishal Chakravarty 氏は、英国を拠点とする医薬品流通企業 NovaPharm Healthcare Ltd(www.novapharmhealthcare.com)の創業者兼CEOです。同社は、ポスト・ブレグジット環境における並行輸入(パラレルインポート)と国境を跨ぐ市場アクセスに焦点を当てています。
同氏は、英国—EU 間の医薬品流通に関する規制コンプライアンス戦略を専門としており、MHRA のパラレルインポート・ライセンス制度、GDP(Good Distribution Practice:医薬品適正流通基準)、および二分化した規制体系下でのサプライチェーン最適化に精通しています。
業務の重点は、分岐した規制体系における実務的な規制適用に置かれています。
英国—EU の規制分岐および並行輸入に関する考察を、vishal.novapharmhealthcare.com で発信しています。
Series Introduction(連載イントロダクション)
ブレグジット後、英国とEUの医薬品規制体系の二分化は、両市場で事業を行う製薬企業の市場アクセス経路を根本的に再構成しました。本連載(全4回)は、ポスト・ブレグジットの規制環境を検討し、二重の規制体系をナビゲートするための実務的示唆を提供します。
- MHRA–NICE Aligned Procedure を含む、英国固有の規制ルートがもたらす戦略的優位性
- 規制分岐がコンプライアンスに与える影響
- ウィンザー・フレームワーク下における並行輸入の実務フレームワーク
- 国境を跨ぐ市場参入に向けた戦略的意思決定
Series Title(連載タイトル)
UK–EU Pharmaceutical Market Access and Compliance in the Post-Brexit Era(ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス)
Articles(各回タイトル)
- Article 1 ブレグジット後の英国・EUにおける医薬品市場アクセスのルート
- Article 2 ブレグジット後の規制およびコンプライアンスの考慮事項
- Article 3 並行輸入の制度枠組みとリスクに関する考察
- Article 4 国境を跨ぐ市場参入に向けたコンプライアンス主導のアプローチ
目次
- 【ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス】第1回 ブレグジット後の英国・EUにおける医薬品市場アクセスのルート Vishal Chakravarty
- 【ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス】第2回 ブレグジット後の規制およびコンプライアンスの考慮事項 Vishal Chakravarty
- 【ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス】第3回 並行輸入の制度枠組みとリスクに関する考察 Vishal Chakravarty