By Vishal Chakravarty
Brexit did not eliminate pharmaceutical regulation. It duplicated it.
On January 1, 2021, the UK pharmaceutical industry transitioned from a unified European regulatory framework to a bifurcated compliance environment requiring parallel systems for UK and EU operations. The Windsor Framework, implemented January 1, 2025, resolved certain Northern Ireland-specific complications but did not reverse the fundamental reality: companies operating across UK and EU markets now maintain dual regulatory infrastructure.
This is not administrative overhead. This is structural cost that reshapes competitive dynamics in European pharmaceutical distribution.
The Compliance Shift: From Shared to Separate Systems
Pre-Brexit, UK pharmaceutical companies operated within EU regulatory harmonization. Good Manufacturing Practice inspections, batch release procedures, pharmacovigilance reporting, and supply chain documentation followed unified standards administered across member states. A UK-based Qualified Person could certify batches for EU distribution. A single Pharmacovigilance System Master File served both jurisdictions. Supply chain traceability systems interfaced with pan-European repositories.
Post-Brexit, this integration dissolved. The UK became a ‘third country’ from the EU regulatory perspective, triggering distinct compliance obligations that now operate in parallel.
Qualified Person Requirements: Geographic Separation
One of the most operationally significant changes involves Qualified Person certification for batch release.
Under EU regulations, pharmaceutical batches destined for EU markets must be certified by a QP physically located within an EEA member state. UK-based QPs can no longer perform this function for EU distribution. Conversely, UK regulations now require UK-based QP certification for batches released into UK markets.
This geographic separation creates several operational implications:
For UK companies distributing to EU: Establishment of QP capacity within an EEA member state became mandatory. This requires either relocating QP operations, contracting with EU-based contract manufacturing organizations, or establishing subsidiary operations with appropriate GMP-certified facilities and qualified personnel.
For EU companies distributing to UK: Similar requirement in reverse. EU QPs cannot certify batches for UK release. Companies must either establish UK QP capacity or engage UK-based partners with appropriate certifications.
For clinical trials: Starting January 2022, investigational medicinal products crossing into Great Britain clinical sites require full UK QP oversight. UK-based QPs can no longer certify clinical trial batches for EEA, necessitating substantive process changes for trials spanning both jurisdictions.
The timeline constraint for establishing this dual QP infrastructure was limited. Where QP certification arrangements were not updated by the end of the transition period (31 December 2020), companies could face significant compliance constraints and potential disruption to lawful batch release for the affected market(s), depending on the product and pathway.
Batch Testing and Release Protocols
Closely related to QP requirements are batch testing location and release procedures.
Under previous arrangements, pharmaceutical manufacturers could conduct batch testing at UK sites listed in Marketing Authorizations and rely on UK QP confirmation for EU release. This reciprocal recognition ended with Brexit.
Current framework requires:
For EU markets: Batch testing must occur at sites located within EEA countries or at facilities covered by Mutual Recognition Agreements that UK no longer holds. If full testing occurs at a UK site still somehow listed in EU Marketing Authorizations (from transitional arrangements), import testing into EU is required. EU QPs cannot rely on UK QP confirmation alone unless a specific trade agreement provision permits this—such provisions remain limited.
For UK markets: Symmetric requirements apply. Batch testing and QP certification must meet MHRA standards with UK-based oversight.
This creates duplication: the same pharmaceutical product targeting both markets may require testing protocols at two separate geographic locations, with independent QP certification processes for each jurisdiction (see Figure 1).
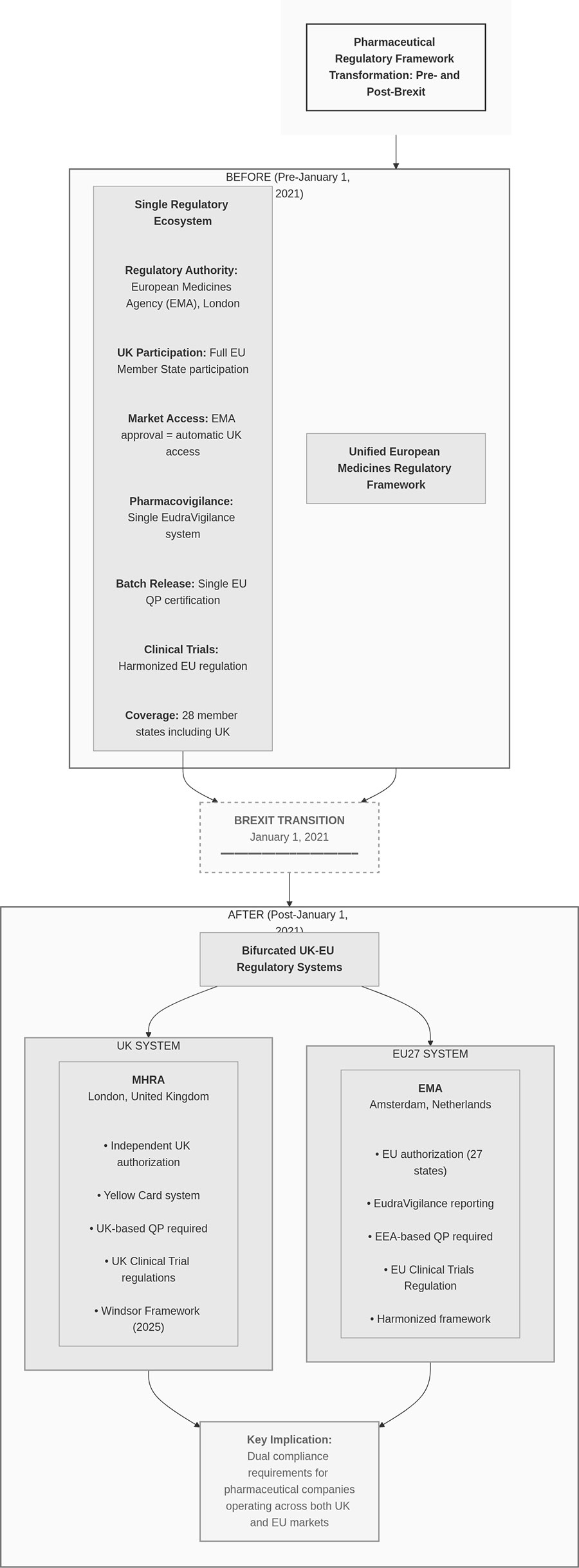
Figure 1. Before/After Framework Comparison (compliance operations)
* Click the image to enlarge
Pharmacovigilance System Separation
Pharmacovigilance infrastructure underwent similar bifurcation.
Pharmacovigilance System Master File (PSMF):
All UK Marketing Authorizations (including Great Britain and Northern Ireland products) must maintain a PSMF available upon MHRA request. While a single PSMF can cover all UK-authorized products, the file must be accessible electronically at the same point in the UK where suspected adverse reactions are accessible. For each UK PV system, a unique UK PSMF number is issued by MHRA following submission; processing timelines may vary.
For products covering Northern Ireland specifically, the PSMF must be located either at the site in EU where main pharmacovigilance activities are performed or where the Qualified Person for Pharmacovigilance operates, due to Northern Ireland’s unique status under Windsor Framework.
Every pharmacovigilance system requires a unique UK PSMF number registered with MHRA.
UK Qualified Person for Pharmacovigilance (UK QPPV):
The UK QPPV role follows requirements similar to EU Good Pharmacovigilance Practices, including medical qualification or access to medical expertise. The UK QPPV can reside in either UK or EEA but holds formal responsibility for UK nationally authorized products. Legal responsibility remains with the UK Marketing Authorization Holder.
Adverse Event Reporting:
UK adverse event reports now flow to MHRA’s Yellow Card system rather than EudraVigilance. Companies operating in both jurisdictions maintain dual reporting systems, with separate data flows, distinct timelines, and independent signal detection processes.
This separation requires clear data governance: safety data for the same product must be properly segregated when reported to UK versus EU authorities while maintaining sufficient integration for global safety surveillance.
Falsified Medicines Directive: Disapplication in UK
A significant regulatory divergence implemented January 1, 2025 involves the EU Falsified Medicines Directive safety features.
Pre-Windsor Framework: FMD applied to prescription-only medicines in EU and Northern Ireland, requiring 2D barcodes containing unique identifiers and anti-tamper packaging. These identifiers were scanned throughout the supply chain and decommissioned upon patient supply through national repository systems.
Post-Windsor Framework: As of January 1, 2025, FMD no longer applies anywhere in UK, including Northern Ireland. UK medicines must not display FMD-compliant features. Where 2D barcodes or serial numbers appear on packaging, they must not be recognized by European repository systems. Any FMD-compliant codes present must be fully removed or covered.
For clarity: the key requirement is that EU FMD Unique Identifiers uploaded to the European repositories system must be removed/covered and must not be EU‑recognisable.
This creates a labeling divergence: products destined for EU markets require FMD features; products for UK markets must not carry functional FMD compliance. Anti-tamper packaging remains encouraged in UK but is not mandated under FMD framework (see Figure 2).
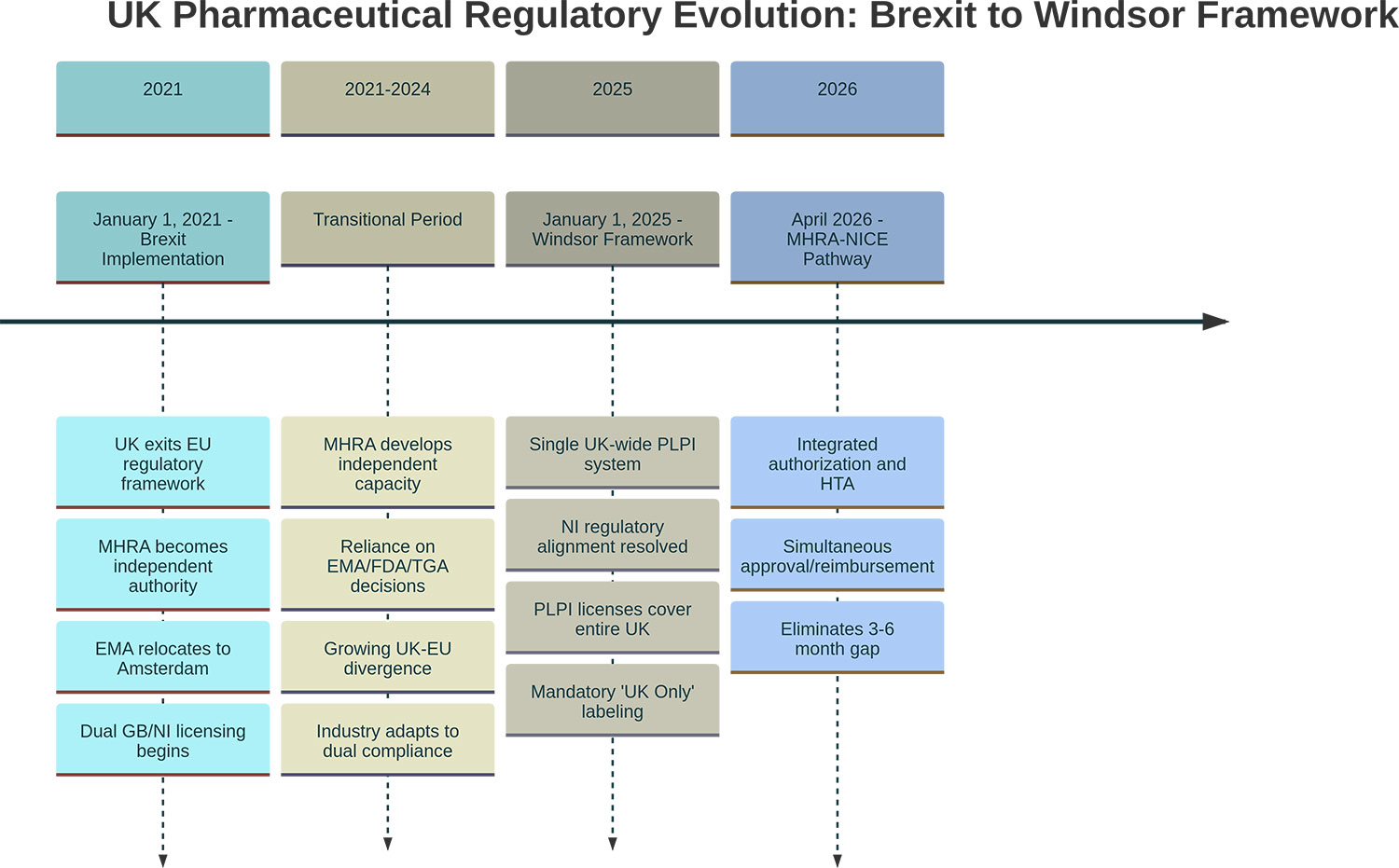
Figure 2. Brexit-to-Windsor Timeline (regulatory milestones)
* Click the image to enlarge
Note: EMA physically relocated from London to Amsterdam in March 2019.
Note: The MHRA–NICE ‘Aligned Pathway’ began early access in Oct 2025; a fully integrated joint scientific advice service is planned by Apr 2026.
Labeling Requirements: ‘UK Only’ Marking
Beginning January 1, 2025, all medicines distributed in UK must carry ‘UK Only’ labels. This requirement applies to all medicines entering UK markets, including parallel imports, wholesale distribution, and domestically manufactured products.
The labeling requirement serves multiple functions: clear market segregation between UK and EU distribution channels, prevention of inadvertent cross-border movement where regulatory status differs, and visible compliance with Windsor Framework provisions.
Wholesalers importing goods through revised parallel import processes must ensure products QP-certified after December 31, 2024 comply with MHRA guidance and feature ‘UK Only’ marking. Packaging printers must be verified for Windsor Framework compliance.
Good Distribution Practice: Documentation and Traceability
While UK Good Distribution Practice standards remain aligned with EU GDP guidelines, operational compliance now occurs under separate regulatory oversight.
UK GDP Oversight: MHRA administers GDP in UK independently. Unlike EU processes, MHRA issues GDP certificates only after inspectors review and accept the distributor’s response to post-inspection letters. This creates a slightly different certification timeline and process.
Documentation Requirements: GDP compliance under both UK and EU frameworks requires comprehensive documentation throughout supply chains: temperature monitoring records with continuous logging, shipping method documentation and transport validation, supplier qualification records and audit trails, traceability systems enabling product tracking from manufacturer to patient, and quality defect handling procedures with root cause analysis.
For companies distributing across both jurisdictions, this means maintaining documentation systems that satisfy both MHRA and EU competent authority requirements, which while similar in principle may have jurisdiction-specific format or retention requirements.
Audit Readiness: UK-site QP sign-offs, secure documentation accessibility, and traceability systems must be audit-ready for MHRA inspections operating under UK-specific timelines and procedures separate from EU GDP inspection regimes.
Supply Chain Operational Adjustments
The compliance separation created practical supply chain challenges requiring operational adjustments:
Inventory Management: Products destined for UK versus EU markets must be managed separately due to labeling differences (UK Only marking, FMD feature presence/absence). This reduces supply chain flexibility and increases working capital requirements as inventory cannot freely interchange between markets.
Quality Control Systems: Separate quality documentation flows for UK and EU QP certification processes require parallel quality management systems or carefully segregated processes within unified systems.
Regulatory Intelligence: Companies must monitor both MHRA and EMA regulatory updates independently. Regulatory changes no longer occur in lockstep; MHRA can diverge when UK-specific considerations warrant different approaches, as seen with decentralized manufacturing regulations implemented July 2025.
Supplier Qualification: Third-country suppliers (outside UK and EU) may require separate qualification processes for UK versus EU distribution channels if different regulatory standards apply.
Cost Structure Implications
The shift from unified to bifurcated compliance creates structural cost increases across several dimensions:
Personnel: Dual QP capacity, separate QPPV arrangements, and independent regulatory affairs teams for MHRA versus EMA interactions.
Facilities: QP certification requires physical presence in jurisdiction, necessitating facility establishment or third-party partnerships in both UK and EEA.
Systems: Separate pharmacovigilance databases, parallel GDP documentation systems, and distinct quality management workflows.
Labeling and Packaging: Separate production runs or post-manufacturing modifications to accommodate UK Only marking and FMD feature requirements.
These costs scale differently depending on company size. Large multinational pharmaceutical companies absorb these costs more easily across high-volume operations. Small and medium-sized enterprises, particularly those focused on niche or specialty medicines, face proportionally higher compliance costs that may affect market entry decisions.
This cost differential creates a structural advantage for larger organizations with resources to maintain parallel regulatory infrastructure, while potentially discouraging smaller operators from serving both markets simultaneously.
Positioning for Long-Term Compliance
The UK regulatory environment continues evolving independently. MHRA can now implement regulatory innovations without EU consensus, as demonstrated by the MHRA-NICE Aligned Pathway and decentralized manufacturing provisions introduced July 2025.
This independent evolution means ongoing regulatory divergence rather than convergence. Companies operating across both markets require sustained regulatory intelligence capabilities, adaptive compliance systems that can accommodate jurisdiction-specific changes, and strategic decisions about which innovations to adopt when UK and EU requirements differ.
Organizations that strengthen internal quality systems, invest in regulatory expertise for both jurisdictions, and build flexible compliance infrastructure that can adapt to independent regulatory evolution will be better positioned for sustained operations in this bifurcated environment.
The compliance considerations post-Brexit are not temporary transitional challenges. They represent permanent structural changes to European pharmaceutical regulatory landscape.
Author Profile Information

Name: Vishal Chakravarty
Title: Founder & CEO, NovaPharm Healthcare Ltd
Contact (E-mail): vishal@novapharmhealthcare.com
Company Website: https://novapharmhealthcare.com/
Writing Portfolio: https://vishal.novapharmhealthcare.com/
Bio
Vishal Chakravarty is Founder and CEO of NovaPharm Healthcare Ltd (www.novapharmhealthcare.com), a UK-based pharmaceutical distribution company focused on parallel import operations and cross-border market access in post-Brexit regulatory environments.
He specializes in regulatory compliance strategy for UK-EU pharmaceutical distribution, with expertise in MHRA parallel import licensing frameworks, Good Distribution Practice requirements, and supply chain optimization under bifurcated regulatory systems. His work focuses on practical applications of pharmaceutical regulatory frameworks in cross-border operations.
He writes on UK-EU pharmaceutical regulatory divergence and parallel import operations at vishal.novapharmhealthcare.com.
Series Introduction
“The bifurcation of UK and EU pharmaceutical regulatory systems following Brexit has fundamentally reshaped market access pathways for pharmaceutical companies operating across both markets. This four-part column examines the post-Brexit regulatory landscape: the strategic advantages of UK-specific pathways including the newly integrated MHRA-NICE Aligned Procedure, the compliance implications of regulatory divergence, practical frameworks for parallel import operations under the Windsor Framework, and strategic decision-making for cross-border market entry. Readers will gain actionable insights into navigating dual regulatory systems and identifying market access opportunities in an increasingly fragmented European pharmaceutical environment.”
Series Title
“UK–EU Pharmaceutical Market Access and Compliance in the Post-Brexit Era”
- Article 1 UK and EU Pharmaceutical Market Access Pathways After Brexit
- Article 2 Regulatory and compliance considerations post-Brexit
- Article 3 Parallel import frameworks and risk considerations
- Article 4 Compliance-driven approaches to cross-border market entry
contents
- 【UK–EU Pharmaceutical Market Access and Compliance in the Post-Brexit Era】1.UK and EU Pharmaceutical Market Access Pathways After Brexit
- 【UK–EU Pharmaceutical Market Access and Compliance in the Post-Brexit Era】2.Regulatory and Compliance Considerations Post-Brexit
- 【UK–EU Pharmaceutical Market Access and Compliance in the Post-Brexit Era】3.Parallel Import Frameworks and Risk Considerations