筆者:Vishal Chakravarty
並行輸入は、競争法と医薬品規制の交差点に位置する仕組みです。認可を受けた流通事業者が、ある正規市場から医薬品を調達し、同じ製品の価格がより高い別の市場で、品質・安全基準を維持する規制枠組みのもとで合法的に流通させることを可能にします。
この仕組みは、経済的効率性と運用上の複雑さの双方を生み出します。同一の医薬品でも、品質の違いではなく価格制度の差によって、ある法域では別の法域より大幅に高価格となることがあります。一方で、並行輸入は、従来の医薬品流通にはない追加のサプライチェーン工程、規制上の監督要件、リスクへの曝露を伴います。
並行輸入の法的枠組み:MHRAの並行輸入ライセンス
並行輸入は、明示的な規制ライセンスを要する特定の法的構成のもとで運用されます。英国では、MHRA(Medicines and Healthcare products Regulatory Agency)がParallel Import Licence制度を管理しています。並行輸入によりUK市場に入る製品は、当該医薬品を対象とする有効なPLPI(Parallel Import Licence)をMHRAから取得していなければなりません。
根本的な要件は、輸入製品が、すでにUKで承認されている製品と治療学的に同等であることを示す点にあります。並行輸入品は、通常の販売承認のようなフルレビューを受けるわけではありません。その代わり、既存のUK販売承認を参照し、輸入品が本質的に同一の医薬品であり、同等の基準で製造されていることを示します。
MHRAにおける並行輸入ライセンス(PLPI)のカテゴリー
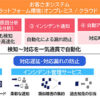
MHRAは、PLPI申請を3つのカテゴリーに分類しています。
シンプルPLPI:UKの販売承認保有者とEEA側の供給元製品の承認保有者が共通の資本関係にある、または明示的なライセンス契約を有する場合に適用されます。必要書類は、企業間の関係性の立証と製品同等性の証明に重点が置かれます。同一企業グループまたは正式なライセンシーが両製品を管理している場合は規制上のリスクが低いため、審査期間は比較的短くなります。承認は、実務上の経験に基づけば、おおむね60〜90日程度となる場合がありますが、個別案件により変動し得ます。
スタンダードPLPI:UK製品と輸入製品に共通起源がないものの、規制上の評価が比較的ストレートに行える場合に適用されます。企業の所有関係が異なっていても、治療学的同等性と同等のGMP基準で製造されていることを示す、包括的な品質関連文書が求められます。
コンプレックスPLPI:共通起源が存在せず、さらに追加的な規制審査が必要となる申請に適用されます。製剤差、製造工程の相違、品質システム上の論点などがある場合、広範な比較評価が必要となり、求められる分析の深さに応じて審査期間も長くなります。

Figure 1. MHRAにおける並行輸入ライセンス(PLPI)のカテゴリー
※画像クリックで拡大表示
※図中の期間は業界実務に基づく目安であり、案件の複雑性や審査内容によって変動し得ます。
ウィンザー・フレームワーク実施による構造変化
2025年1月1日は、ウィンザー・フレームワークの実施により、英国の並行輸入制度に大きな変化をもたらした節目となりました。
▽対象地域の拡大:すべての並行輸入ライセンスは、北アイルランドを含む英国全域を対象とするようになりました。MHRAは、4つの構成国すべてにおけるUK並行輸入の唯一のライセンス当局となり、既存のGB限定PLPIは、新規申請や追加費用なしで自動的にUK-wide認可へ切り替えられました。
▽偽造医薬品指令(FMD)の適用除外:EUのFMD要件は、UK向け並行輸入には適用されなくなりました。並行輸入によりUK市場へ入る製品は、機能するFMD準拠の特徴を表示してはなりません。包装上にEU規則2016/161に基づく英数字シーケンスを符号化した2Dバーコードが存在する場合は、完全に除去または被覆する必要があります。
▽表示要件:「UK Only」:すべての並行輸入製品には「UK Only」表示が必要です。これにより、UK向け製品であることを明確化し、異なる規制要件が適用される越境移動を防ぎます。
サプライチェーン上のリスクに関する考察
並行輸入は、従来の医薬品流通では回避されるか、別の形で管理されるリスク類型を生み出します。
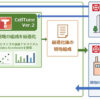
▽サプライチェーンの複雑化:従来の流通は、メーカー→正規卸→調剤薬局という比較的単純な経路です。これに対し並行輸入では、EEAメーカー→EEA卸→並行輸入事業者→UK卸→調剤薬局という工程が加わります。工程が増えるごとに、製品の完全性、保管条件、文書の正確性、トレーサビリティが損なわれる潜在的なポイントが増えます。
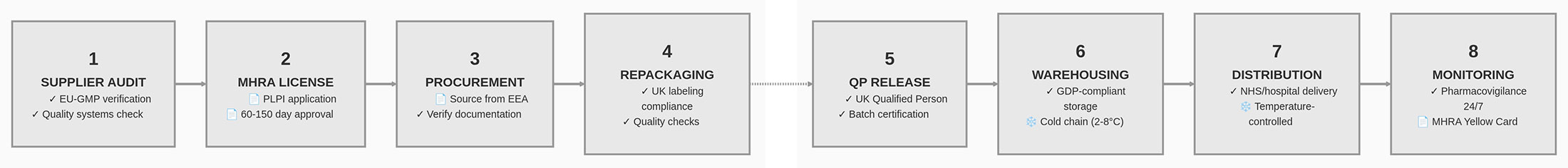
Figure 2. 並行輸入におけるサプライチェーン工程
※画像クリックで拡大表示
▽再包装・再表示のリスク:多くの並行輸入品は、UK表示要件に適合させるため、再包装や再表示が必要になります。これにより、再包装工程での製品識別の正確性、保管条件の維持、表示情報の正確な転記、EEAの元包装からUK最終流通までのバッチトレーサビリティ維持といった運用リスクが生じます。
再包装を行う企業は、組立工程をカバーする適切な製造業許可を保有していなければなりません。また、再包装工程が元の製造と同等のGMP基準を満たすことを保証する品質システムが必要です。
▽製品認証と偽造リスク:並行輸入のサプライチェーンは、越境移動と複数の仲介者を伴うため、偽造品や不正品が入り込む潜在的な入口を生みます。並行輸入経路を通じた偽造品混入は、認識されたリスクの一つであり、回収を含むMHRAの執行措置は、英国の供給網における製品認証上の不備に対して歴史的に講じられてきました。
これを低減するには、強固な供給者適格性確認プロセス、文書とは独立して製品の真正性を確認するバッチ検証システム、さらに外観・包装・文書上の異常を継続的に監視する仕組みが必要です。
▽規制上の責任:並行輸入では、PLPI保有者が、自ら製造していない製品についても規制上の責任を負う構造になります。品質問題、有害事象、規制不適合が並行輸入品で発生した場合、ライセンス保有者はMHRAに対する説明責任を負います。この責任は、PLPI保有者が直接管理していない品質システムや仕様のもとで製造された製品にも及びます。
品質保証上の要件
MHRAは、並行輸入事業者に対して、複数の側面をカバーする堅牢な品質システムを求めています。
▽供給者適格性確認:EEA供給者について、GMP適合性の確認、卸売業に関するGDP認証の確認、供給者の品質マネジメントシステムの評価、継続的なパフォーマンス監視などを、文書化された形で運用する必要があります。
▽バッチリリース手順:実務上、各並行輸入バッチは、特に再包装や組立工程を伴う場合、UK流通前にQP認証が必要となるのが一般的です。QPは、製品が品質規格を満たしていること、サプライチェーン全体で保管条件が適切だったこと、文書により正しい受渡し履歴が確認できること、再包装・再表示が適切な基準で実施されたことを確認しなければなりません。
▽保管・輸送のバリデーション:温度管理が重要な医薬品では、国際輸送中の温度モニタリング、各拠点の妥当性確認済み保管設備、逸脱時対応手順など、並行輸入全体を通じたコールドチェーン管理のバリデーションが必要です。
▽ファーマコビジランス体制:PLPI保有者は、並行輸入製品について適切なPV体制を維持する必要があります。有害事象報告の監視、MHRAとの安全性シグナル対応、品質問題発生時の製品回収対応、そして回収が必要になった場合に迅速にバッチ所在を特定できるトレーサビリティ体制が含まれます。
経済的リスク要因
規制・品質リスクに加えて、並行輸入には事業性に影響する経済面の考慮事項もあります。
▽価格差の変動性:並行輸入の採算は、供給元市場と販売先市場の価格差が継続することに依存します。しかし、この差は、メーカーの価格調整、為替変動、各国の償還制度変更、いずれかの市場における競争環境の変化などによって縮小または消失する可能性があります。
▽規制コスト負担:MHRAはPLPI申請や変更申請に手数料を課しています。品質システムの維持、QPサービス、GDP対応インフラ、PV体制などは継続的な運用コストを伴います。低数量・低マージン製品では、こうした固定コストが価格差による利益を上回る可能性があり、十分な数量と価格差が見込める製品に対象が絞られる要因になります。
▽市場参入の不確実性:Simple PLPIでは比較的予見可能であっても、MHRAの承認期間は、調達機会を見つけてから実際に市場アクセスを得るまでの遅延要因になります。審査中に市場環境が変わる、供給元製品が入手困難になる、UKの参照製品に変更が生じるといったリスクもあります。
リスク低減のための対応策
並行輸入に取り組む組織は、これらのリスクに対応するために、いくつかの実務的アプローチを採ることができます。
・強固な供給者適格性確認プログラム:可能な場合の現地監査、第三者によるGMP・GDP適合性確認、責任分担を明示した品質契約、逸脱時のエスカレーション手順を含む継続的パフォーマンス監視。
・包括的品質システム:調達から最終流通まで、並行輸入業務の全工程をカバーする文書化された手順、定期内部監査、継続的改善プロセス。
・保守的な製品選定:共通起源のある案件(Simple PLPI)、規制投資を正当化できる安定した価格差、安全性プロファイルが確立している製品、固定規制コストを吸収できる十分な数量ポテンシャルを持つ案件に重点を置くこと。
構造的な所見
並行輸入の制度は、裁定取引を通じた市場効率性の実現と、医薬品の品質・安全性の維持という相反する目標のバランスを取っています。規制枠組みは、治療学的同等性の確認と、並行輸入事業者に対する品質システム要件を通じて、その均衡を図っています。
そのリスクプロファイルは、従来の医薬品流通とは明確に異なります。追加的なサプライチェーンの複雑性、再包装工程、越境規制調整、そして無関係の製造主体が作った製品に対する責任負担など、専門的な管理能力を要する曝露が存在します。
ウィンザー・フレームワークの実施により、UK-wide の対象地域化や、GB/NI二重ライセンス要件の撤廃といった一部の運用は簡素化されました。しかし、サプライチェーンの完全性、製品認証、品質システムの堅牢性、経済的な成立性といった、並行輸入に固有の基本リスクは引き続き残ります。
並行輸入事業に参入する組織には、規制上の専門性、洗練された品質システム、そしてリスク管理の規律が求められます。この制度は、従来流通よりも複雑なサプライチェーン全体にわたり医薬品の安全性を維持できるだけの運用品質を前提として、この流通モデルを認めているのです。
Author Profile(著者プロフィール)

氏名(Name):Vishal Chakravarty
役職(Title):Founder & CEO, NovaPharm Healthcare Ltd
連絡先(E-mail):vishal@novapharmhealthcare.com
企業ウェブサイト:https://novapharmhealthcare.com/
執筆ポートフォリオ:https://vishal.novapharmhealthcare.com/
Bio(略歴)
Vishal Chakravarty 氏は、英国を拠点とする医薬品流通企業 NovaPharm Healthcare Ltd(www.novapharmhealthcare.com)の創業者兼CEOです。同社は、ポスト・ブレグジット環境における並行輸入(パラレルインポート)と国境を跨ぐ市場アクセスに焦点を当てています。
同氏は、英国—EU 間の医薬品流通に関する規制コンプライアンス戦略を専門としており、MHRA のパラレルインポート・ライセンス制度、GDP(Good Distribution Practice:医薬品適正流通基準)、および二分化した規制体系下でのサプライチェーン最適化に精通しています。
業務の重点は、分岐した規制体系における実務的な規制適用に置かれています。
英国—EU の規制分岐および並行輸入に関する考察を、vishal.novapharmhealthcare.com で発信しています。
Series Introduction(連載イントロダクション)
ブレグジット後、英国とEUの医薬品規制体系の二分化は、両市場で事業を行う製薬企業の市場アクセス経路を根本的に再構成しました。本連載(全4回)は、ポスト・ブレグジットの規制環境を検討し、二重の規制体系をナビゲートするための実務的示唆を提供します。
- MHRA–NICE Aligned Procedure を含む、英国固有の規制ルートがもたらす戦略的優位性
- 規制分岐がコンプライアンスに与える影響
- ウィンザー・フレームワーク下における並行輸入の実務フレームワーク
- 国境を跨ぐ市場参入に向けた戦略的意思決定
Series Title(連載タイトル)
UK–EU Pharmaceutical Market Access and Compliance in the Post-Brexit Era(ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス)
Articles(各回タイトル)
- Article 1 ブレグジット後の英国・EUにおける医薬品市場アクセスのルート
- Article 2 ブレグジット後の規制およびコンプライアンスの考慮事項
- Article 3 並行輸入の制度枠組みとリスクに関する考察
- Article 4 国境を跨ぐ市場参入に向けたコンプライアンス主導のアプローチ
目次
- 【ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス】第1回 ブレグジット後の英国・EUにおける医薬品市場アクセスのルート
- 【ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス】第2回 ブレグジット後の規制およびコンプライアンスの考慮事項
- 【ポスト・ブレグジット時代における英国・EUの医薬品市場アクセスとコンプライアンス】第3回 並行輸入の制度枠組みとリスクに関する考察